
Enzymes have clearly defined active sites to allow the substrate molecule to fit intricately. This is often coupled with an enzymatic conformational change prior to the occurrence of the catalysis reaction. For Ago, the catalysis step requires insertion of a “glutamate finger” to form the catalytic plugged-in conformation, which can be stabilized through hydrogen-bonding networks provided by two symmetric positively-charged residues.
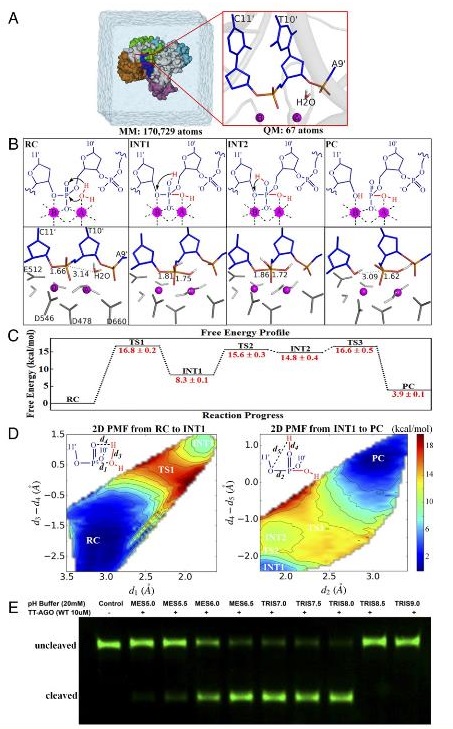
Figure : The substrate-assisted target DNA cleavage mechanism. (A) Overview of the setup used for our simulations forTtAgo-mediated target DNA cleavage.Snapshot of the simulation box (Left) and detailed view of the QM subsystem in the precleavage state (Right; PDB ID code 4NCB). (B) Detailed cleavagemechanism (Top) and respective critical structures (Bottom).Movies S1andS2show the mechanism details. The coordinating amino acid residues D546, D478,D660, and E512 are colored dark gray, and the coordinating waters that do not participate in the cleavage reaction are colored light gray. INT1, first in-termediate; INT2, second intermediate; PC, product complex, i.e., postcleavage state; RC, reactant complex, i.e., precleavage state; TS1, first transition state;TS2, second transition state; TS3, third transition state. (C) Calculated free energy changes with their statistical errors along the reaction progress (in kilo-calories per mole). Approximately 430 windows were selected to perform a total of approximately 6.5ns B3LYP(6-31G*) QM/MM-MD simulations to generatethe free energy profile. (D) Calculated 2D PMF along the reaction progress (in kilocalories per mole). The reaction coordinated1involves the nucleophilicattack of water to form a new P–O bond, reaction coordinated3−d4is the hydrogen transfer from nucleophilic water to the pro-Rp phosphate oxygen oftarget nucleotide T10′, reaction coordinated2is the breaking of P–O bond between target nucleotides T10′and C11′, and reaction coordinated4−d5is theproton transfer from the pro-Rp oxygen of nucleotide T10′to the 3′leaving oxygen of target nucleotide C11′.(E) pH-dependent experimental measurementsfor the cleaved product of WT .
For Ago in eukaryotes, these two symmetric positively-charged residues play the identical role that is critical for cleavage. Hence, it was long speculated that the two analogous resides in prokaryotic Ago perform the same critical role in cleavage function. Surprisingly, this study showed that in pAgo, only one (Arginine 545) of the two residues is involved in cleavage function. When the other one (Arginine 486) was substituted with other amino acids, the enzyme was still able to maintain its cleavage activity. Based on these results, the study further suggested that R486 may play other roles such as assisting the insertion of the glutamate finger. The discovery of such striking differences in the roles of these symmetric resides between eAgos and pAgos provides novel insights on how the cleavage functions evolve during the evolution journey from prokaryote to eukaryote.
To achieve these results, computational methods combining Quantum Mechanics, Molecular Mechanics, and Molecular Dynamics (QM/MM) were applied to elucidate the cleavage reaction mechanism and identify functional roles of the amino acid residues. This research was made possible by large-scale high-performance computing resources, which were computed equivalent to 10,000 CPU cores for 25 weeks on the Shaheen II Supercomputer at KAUST in collaboration with Prof. Xin GAO’s group.
“This research was made possible due to current day computing capabilities and the precision that QM/MM modelling allows for,” said Prof. HUANG Xuhui. “Comparing which amino acid residues play a key part in the target DNA/RNA cleavage step in pAgo and eAgo sheds light on how Ago protein evolves from prokaryotes to eukaryotes to cleave DNA/RNA. This information may be useful in ultimately modifying the Ago protein for use as an enhanced gene editing tool in the future,” Prof. Huang explained.
Source :www.sciencedaily.com

























